Patients demonstrate autoantibodies directed against type VII collagen, a major component of the skin's basement membrane. The most common antigenic target is the noncollagenous1 (NC-1) portion of type VII collagen. Most commonly, the antibodies are of the immunoglobulin G (IgG) subtype, but IgA, IgE, and IgM subtypes have also been reported.
There are several clinical forms of EBA, as defined by the International Bullous Diseases Group:
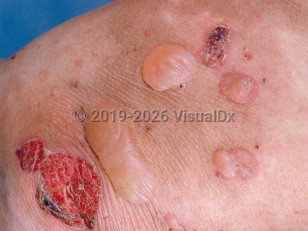
Classical / Mechanobullous form: A noninflammatory form that affects trauma-prone sites and extensor skin surfaces.
Nonclassical / Nonmechanobullous (inflammatory) forms:
- Bullous pemphigoid (BP)-like – An inflammatory form with features characteristic of BP mixed with atypical lesions for BP, such as skin fragility and milia. It is characterized by a generalized eruption of vesicles and bullae.
- Mucous membrane pemphigoid-like – A mucous membrane pemphigoid-like form, which predominantly affects mucous membranes (mouth, pharynx, conjunctiva, genitalia).
- Brunsting-Perry pemphigoid-like – A chronic, recurrent blistering dermatosis of the head and neck that presents with scarring alopecia.
- Linear IgA dermatosis-like – A presentation that may resemble linear IgA bullous dermatosis and presents with linear IgA deposits in the basement membrane.
EBA can be associated with inflammatory bowel disease (IBD), especially Crohn disease. As many as 25% of EBA patients in the United States have IBD, but this number varies greatly between studies. It is postulated that the damaged gut epithelium exposes previously sequestered collagen VII to the immune system, resulting in the generation of autoantibodies that escape and attack the skin. EBA has been also reported to be a paraneoplastic phenomenon, and in association with psoriasis vulgaris, thyroiditis, chronic lymphoid leukemia, diabetes mellitus, rheumatoid arthritis, multiple endocrinopathy syndrome, and amyloidosis. A meta-analysis estimated the prevalence of associated comorbidities at close to 10%.
Associations with endocrinopathies and other systemic complications do not seem to follow the same rate in pediatric cases. The few pediatric cases that have associated autoinflammation were also found to have HLA-DR2 (most frequently), HLA-DR4, HLA-DR5, or HLA-DR7 aberrancy.
An increased incidence of EBA in patients with darker skin phototypes has previously been noted. This has now been attributed to enriched representation of the HLA-DRB1*15:03 allele found in Americans of African descent, as well as HLADRB1*16.
EBA typically has a slow onset and a chronic clinical course. Scarring may produce secondary dysfunction, particularly of the hands and fingers, in the mechanobullous variant. Other sequelae include hair and nail loss, esophageal stenosis, periodontal disease, malnutrition, and blindness.
Related topic: epidermolysis bullosa simplex