Current thinking suggests that the causative mutation in sickle cell disease primarily arose on the African continent due to the protective effect of the carrier state against malaria, so, globally, most patients share an African ancestry. The disease is most prevalent in Nigeria, where nearly 150 000 children are born with sickle cell disease annually. Additionally, there is a high incidence of sickle cell disease in individuals of Caribbean descent and those from Mediterranean countries such as Turkey, Greece, and Italy. In the United States, about 100 000 people have sickle cell disease.
In the United States, sickle cell disease is the most frequently detected condition in newborn-screening programs, regardless of ethnicity. Originally as a result of the transatlantic slave trade from Africa, nearly all sickle cell disease patients in the United States are Black.
Hemoglobin S has diminished solubility, especially when there is low oxygen tension. This diminished solubility results in the characteristic sickle shape of the affected red blood cells, leading to hemolysis and adherence to vascular endothelium, which leads to vascular occlusion, coagulation activation, and subsequent ischemia and tissue necrosis.
Acute presentations include vaso-occlusive crises (painful episodes of microvascular occlusion that involve joints, bones, and internal organs such as the spleen, liver, kidneys, and central nervous system; see sickle cell acute pain crisis and acute chest syndrome), hematologic crises (due to pooling of blood in the enlarged spleen), and infectious crises (result from functional asplenia, predisposing to infections from encapsulated organisms).
These painful crises cause significant suffering and stigmatization for sickle cell patients, who are often unjustly described as drug seekers and accused of faking their pain. This results in inadequate treatment, as well as added stress and suffering, leading many patients to avoid care altogether. Without adequate treatment, sickle cell disease can affect any organ and is associated with decreased quality of life and a shortened life span.
Chronic clinical manifestations of disease include hemolytic anemia and a variety of systemic symptoms and signs that occur as a result of microvascular occlusion (eg, ischemia, necrosis, splenic autoinfarction). Osteonecrosis occurs in up to one-half of patients with sickle cell disease and can be associated with pain lasting longer than that associated with vaso-occlusive events. If untreated or recurring, osteonecrosis can lead to fractures and osteomyelitis.
Patients with sickle cell disease are at increased risk of venous thromboembolic disease, including recurrently. This risk increases with hospitalizations. Up to 12% of sickle cell patients will have a thrombotic event before age 40. Around a quarter of sickle cell patients will have at least one stroke by their fourth decade of life. Posterior reversible encephalopathy syndrome is a strong mimic of stroke in sickle cell patients.
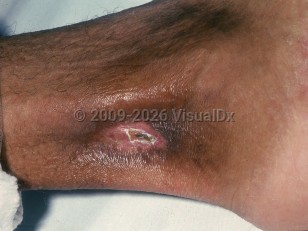
Leg ulcers, which can be extremely painful, are considered a marker of disease severity. The shins, dorsal feet, Achilles tendon, or ankles are frequent sites of involvement, as these are areas with little subcutaneous tissue and thin overlying skin with decreased blood flow. Ulcer formation may be spontaneous or may result from local trauma. Sickle cell ulcers are more common in males.
Factors that predispose to ulcer formation are:
- Vessel obstruction by sickle cells.
- Increased venous and capillary pressure.
- Secondary bacterial infection.
- Decreased oxygen carrying capacity of the blood, leading to peripheral ischemic changes in the skin.

 Patient Information for
Patient Information for