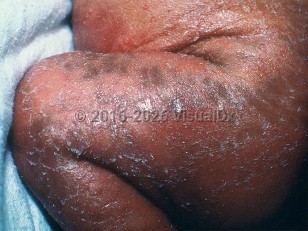
Conradi-Hunermann-Happle syndrome is a rare subtype of chondrodysplasia punctata, a heterogeneous group of skeletal dysplasias. It is caused by a defect in cholesterol synthesis due to a mutation of the emopamil-binding protein (EBP) gene, encoding EBP, and transmitted in a mosaic X-linked dominant fashion. It is characterized by short stature, craniofacial defects, sectorial cataracts, ichthyosis, coarse hair, and alopecia.
Fewer than 100 cases have been reported worldwide. It is presumed lethal in males (although it is rare that males are reported with the syndrome), but affected females have a normal intelligence and life span.
Characteristic skin changes are present at birth and evolve as the child ages. Hair and nail changes occur, but the teeth remain normal. Skeletal anomalies include short stature, shortening of the rhizomelic limbs, epiphyseal stippling, and craniofacial defects.
Eye abnormalities include unilateral sectorial cataracts at birth or soon after, and sometime microphthalmia or microcornea. Occasional problems are congenital heart defects, sensorineural deafness, central nervous system malformations, or renal malformations.
Variable features include rounded or asymmetrical facies with frontal bossing, a broad and flat nasal bridge, shortened limbs, kyphoscoliosis, supernumerary digits, and other skeletal defects. Recurrent flexural skin infections may be problematic.
Conradi disease in Child
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
Q77.3 – Chondrodysplasia punctata
SNOMEDCT:
398958000 – Chondrodysplasia punctata, X-linked dominant type
Q77.3 – Chondrodysplasia punctata
SNOMEDCT:
398958000 – Chondrodysplasia punctata, X-linked dominant type
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Updated:04/09/2023
Conradi disease in Child