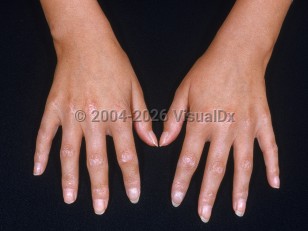
Pediatric scleroderma, or juvenile systemic sclerosis (JSSc), is a rare and severe multisystem autoimmune connective tissue disease that involves sclerotic changes of the skin and internal organs. It is estimated to affect 3 in 1 000 000 children.
While the etiology remains unknown, the disease is characterized by autoantibody production, collagen deposition, and vascular dysfunction. The mean age of onset in childhood is 8 years, and girls are affected more frequently than boys. Exceedingly rarely, disease onset may occur in infancy, when other congenital sclerosing disorders such as restrictive dermopathy should be considered.
Raynaud phenomenon is usually the initial presenting symptom of JSSc, along with nail fold capillary changes and positive antinuclear antibodies (ANA).
There are 3 major clinical subsets of scleroderma that are seen in childhood:
- Limited cutaneous scleroderma – Distal skin sclerosis, Raynaud phenomenon, frequent severe late-stage complications such as pulmonary hypertension, renal crisis, and gastrointestinal involvement. CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasias) refers to a subset of patients with limited scleroderma. CREST syndrome is rare in children.
- Diffuse cutaneous SSc – Proximal extremity or truncal skin demonstrates sclerosis early on; Raynaud phenomenon of shorter duration, early systemic involvement such as cardiac and lung fibrosis, and a high risk of renal crisis. Some patients experience rapidly progressive disease, but most experience a slower, more chronic course.
- Overlap syndrome with scleroderma – In this condition, limited or diffuse scleroderma occurs in concert with another connective tissue disease, such as systemic lupus erythematosus (SLE), juvenile dermatomyositis, or Sjögren syndrome. Overlap syndrome is more common in children than adults.