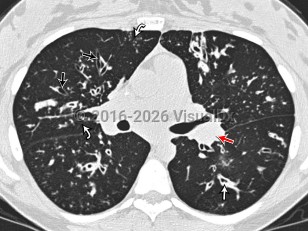
Cystic fibrosis is a congenital metabolic disorder caused by a mutation in a protein that regulates chloride ion transport. This leads to viscous secretions and eventual dysfunction of exocrine glands, most notably affecting the lungs and pancreas. Symptoms usually appear in childhood and include recurrent pulmonary infections with gradual loss of pulmonary function and pancreatic insufficiency leading to poor weight gain. Liver involvement can range from steatosis to advanced cirrhosis with portal hypertension.
Only about 4% of cystic fibrosis patients are diagnosed as adults. Adult patients may also develop pancreatic endocrine insufficiency and cystic fibrosis-related diabetes, which involves both insulin deficiency and resistance. Most adult cystic fibrosis patients will ultimately die of respiratory failure.
Pseudomonas aeruginosa, Staphylococcus aureus, and Mycobacterium abscessus infections are complications of cystic fibrosis. Patients with cystic fibrosis may be anemic. Patients often have impaired absorption in the small bowel and ciliary dysfunction in the genitourinary system that can impact fertility.
Cystic fibrosis is an autosomal recessive disorder with a high prevalence in Ashkenazi Jewish populations. In these high-risk populations, routine genetic testing is often pursued.
Early trials of combining 2 genetic therapies have shown promising results in their potential to treat the underlying genetic cause of disease in the majority of patients with cystic fibrosis. Despite significant improvements in therapy, primarily pertaining to pulmonary function and nutrition, life expectancy is still decreased but has been improving over the past several decades.
Cystic fibrosis in Infant/Neonate
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
E84.9 – Cystic fibrosis, unspecified
SNOMEDCT:
190905008 – Cystic Fibrosis
E84.9 – Cystic fibrosis, unspecified
SNOMEDCT:
190905008 – Cystic Fibrosis
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:01/31/2019
Last Updated:10/13/2024
Last Updated:10/13/2024
Cystic fibrosis in Infant/Neonate