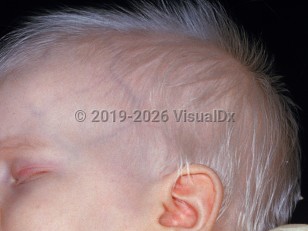
Oculocutaneous albinism (OCA) represents a group of autosomal recessive disorders characterized by dilution or absence of melanin pigment in the skin, hair, and eyes. In all variants of albinism, melanocytes and melanosomes are normal. The reduction or absence of melanin is caused by abnormalities in tyrosinase or enzymes involved in the biosynthesis and distribution of melanin. The worldwide prevalence is estimated at 1:17 000. All patients with albinism have an increased risk of contracting skin cancers.
Eight subtypes of OCA have been recognized, caused by mutations in different genes.
The genes associated with OCA1-8 are listed below:
- OCA1: TYR – tyrosinase
- OCA2: OCA2 – P gene
- OCA3: TYRP1 – tyrosinase-related protein 1
- OCA4: SLC45A2 – solute carrier family 45 member 2
- OCA5: mapped to chromosome 4q24
- OCA6: SLC24A5 – solute carrier family 24 member 5
- OCA7: LRMDA – leucine-rich melanocyte differentiation associated
- OCA8: DCT- Dopachrome tautomerase
OCA1 is the most common subtype found in individuals of Northern European descent (50% of cases). OCA2 accounts for 30% of cases worldwide and is the most common cause of OCA in Africa. OCA3 is virtually unseen in individuals of Northern European descent but accounts for 3% of cases worldwide. OCA4 is rare in individuals of Northern European descent and in African populations, but it accounts for 17% of cases worldwide, especially in Asia.
Pigmentary dilution of skin and hair occurs in all patients, but the degree of hypopigmentation varies with each type of albinism.
Ocular changes of OCA and ocular albinism (OA) include nystagmus, reduced visual acuity, strabismus, iris translucency, absent or reduced pigment of the retinal pigment epithelium, misrouting of the optic nerves, foveal hypoplasia, and photophobia.


 Patient Information for
Patient Information for