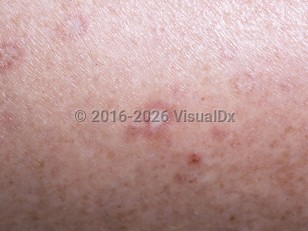
Malignant atrophic papulosis (MAP), or Degos disease, is a rare primary vaso-occlusive disorder presenting on the trunk or extremities as rose-colored papules that become umbilicated, healing with a porcelain white center with surrounding telangiectasia.
The cutaneous and systemic manifestations of MAP may cause significant pain.
The disease is thought to exist in 3 forms. The classic systemic form affects the skin first and later may involve the eyes, gastrointestinal tract, heart, and central nervous system (CNS), generally resulting in death within several years because of sepsis from peritonitis (61%), CNS bleeding (18%), and pleural or pericardial involvement (16%). A benign, purely cutaneous form is seen in up to 15% of patients and is defined by no signs of systemic involvement within 3 years of diagnosis. Finally, MAP may be seen in association with connective tissue diseases such as antiphospholipid syndrome, lupus erythematosus, dermatomyositis, and systemic sclerosis.
MAP typically occurs between the second and fourth decades of life. Cases of MAP have been reported in teenagers and children. The incidence in men may be greater than in women. Although most cases are sporadic, there are reports of familial cases.
The pathogenesis of MAP is unknown, although vascular coagulopathy and vasculopathy are possible pathogenic mechanisms. Other theories implicate a viral or autoimmune pathogenesis. A combination of prothrombotic factors possibly plays a role; however, in studies of MAP patients, no single abnormality was repeatedly identified.
Malignant atrophic papulosis
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
I77.89 – Other specified disorders of arteries and arterioles
SNOMEDCT:
400171002 – Malignant atrophic papulosis
I77.89 – Other specified disorders of arteries and arterioles
SNOMEDCT:
400171002 – Malignant atrophic papulosis
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Updated:01/19/2022
Malignant atrophic papulosis