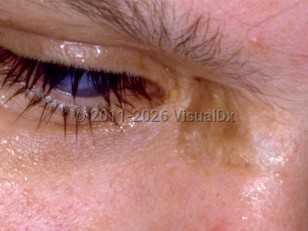
Homocystinuria is an autosomal recessive inherited disorder of methionine amino acid metabolism. Homocystinuria encompasses a group of inborn errors of metabolism characterized by elevated serum and urine homocysteine levels. Homocystinuria presents as a multisystem disease involving connective tissue, the cardiovascular / vascular system, and the central nervous system.
Classic homocystinuria is due to mutations in the cystathionine beta-synthase gene on chromosome 21q22.3 resulting in cystathionine beta-synthase enzyme deficiency. Excess homocysteine is remethylated to methionine, which, along with its metabolites, is toxic to the nervous system. Normal life span may occur with early intervention in vitamin B6 (pyridoxine) responders. Otherwise, life expectancy is reduced from thromboembolic complications (approximately 25%-50% by age 30).
Other types of homocystinuria may result from defects in methionine synthase or methionine synthase reductase and defects in vitamin B12 (cyanocobalamin) metabolism. These patients do not present with increased methionine levels on neonatal screens. However, clinical manifestations are similar to those of classic homocystinuria.
Homocystinuria is relatively rare (approximately 1:300 000 worldwide) with a higher incidence in Ireland. It occurs more often in males than females.
Homocystinuria in Adult
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
E72.11 – Homocystinuria
SNOMEDCT:
11282001 – Homocystinuria
E72.11 – Homocystinuria
SNOMEDCT:
11282001 – Homocystinuria
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Updated:01/17/2022