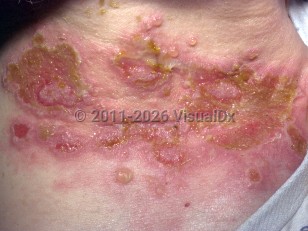
Paraneoplastic pemphigus (PNP), or paraneoplastic autoimmune multiorgan syndrome (PAMS), is a variable multiorgan autoimmune syndrome with severe mucocutaneous disease that typically develops in the setting of current or past history of a lymphoreticular neoplasm, mainly B-cell or thymoma-like neoplasms. It is a rare disease, accounting for 3%-5% of all pemphigus cases.
Most patients (approximately two-thirds) who develop PNP have a preceding history of a neoplasm. In the remaining one-third of patients, the onset of PNP will precede the diagnosis of the malignancy. There are rare reports of no detectable malignancy at the time of the development of PNP. Specific neoplasms associated with PNP include non-Hodgkin lymphoma, chronic lymphocytic leukemia, Castleman tumor (angiofollicular lymph node hyperplasia), thymoma, spindle cell neoplasms, epithelial neoplasms, sarcomas, and Waldenström macroglobulinemia.
The disease is thought to be due to cross-reactivity between tumor antigens and epithelial antigens, causing both mucosal and cutaneous lesions. Immunoglobulin G (IgG) antibodies develop against multiple antigens, such as members of the plakin and desmoglein families: desmoplakin I (DPI1), desmoplakin II (DPI2), plectin, envoplakin, periplakin, bullous pemphigoid antigen 230, desmoglein 1 (DSG1), and desmoglein 3 (DSG3).
The disease is most commonly identified in adults aged 45-70, as this is the demographic most likely to develop lymphoma. Patients with Castleman disease tend to be younger, in the second or third decade of life. There is no association with sex, race / ethnicity, or geographic region, although there may be an association with DRB1*03 and HLA-Cw*14 alleles.
Symptoms and signs include painful cutaneous and oral lesions secondary to the vesicles and bullae that form and subsequently rupture. Severe eye irritation may also be seen with conjunctival involvement, and esophageal, nasopharyngeal, vaginal, and penile mucosal lesions may also be seen. Pulmonary involvement, occurring in around 90% of patients, takes the form of bronchiolitis obliterans, which is frequently fatal.
Prognosis depends on the associated malignancy. Removal of some tumors (thymoma or Castleman disease) may induce disease remission. However, patients with other malignancies may deteriorate, with death most often due to bronchiolitis obliterans, sepsis, gastrointestinal bleeding, or organ failure. Decreased survival has also been noted in patients with a bullous pemphigoid-like and a toxic epidermal necrolysis (TEN)-like picture as well as the presence of bronchiolitis obliterans.
Pediatric patient considerations: Castleman disease is the most commonly associated condition in children.
Paraneoplastic pemphigus
See also in: Oral Mucosal LesionAlerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
L10.81 – Paraneoplastic pemphigus
SNOMEDCT:
402718003 – Pemphigus paraneoplastica
L10.81 – Paraneoplastic pemphigus
SNOMEDCT:
402718003 – Pemphigus paraneoplastica
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:07/29/2024
Last Updated:08/01/2024
Last Updated:08/01/2024
Paraneoplastic pemphigus
See also in: Oral Mucosal Lesion