VKH syndrome results from a T-cell-mediated autoimmune response to melanocytes of central nervous system and skin in genetically susceptible individuals. The event that triggers this response remains unidentified. The syndrome is reported to occur more frequently among people of Asian, American Indian, Hispanic, and Indian descent. The disease more commonly affects women during the third or fourth decade of life. Individuals carrying the HLA-DRB1*0405 allele are most at risk.
VKH syndrome presents in 4 clinically distinct phases:
- Prodromal – Acute onset of fever, headache, nuchal rigidity, tinnitus, vertigo, and hearing loss, which typically last 3-5 days before complete resolution.
- Uveitic – Within a few days, patient develops acute bilateral uveitis, exudative retinal detachment, and optic disc swelling, which present as visual loss, blurring, photophobia, and ocular pain.
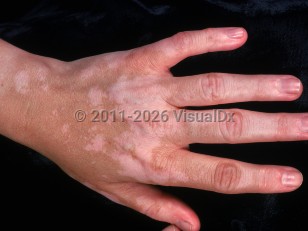
- Convalescent – Within several weeks, patient develops diffuse symmetric depigmentation of the choroids and poliosis, vitiligo, or alopecia that typically affects scalp, face, bilateral eyebrows, and eyelashes, which may last months to years.
- Chronic recurrent – Within months to years, about two-thirds of patients progress to the chronic phase, which is characterized by recurrent episodes of anterior uveitis.
VKH syndrome more frequently affects those in their third or fourth decade; however, there have been reports of VKH syndrome in children.