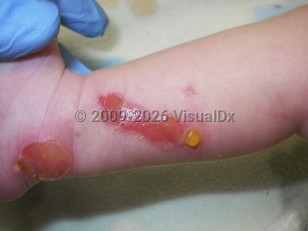
Transient bullous dermolysis of the newborn (TBDN) is a rare form of dystrophic epidermolysis bullosa (DEB) characterized by subepidermal blisters that are either congenital or develop because of friction during the neonatal period. The condition remits spontaneously within a couple of months to the first or second year of age. Vesicles are induced by minimal trauma and can affect extensor surfaces, or they may be more widespread, including mucous membranes and nails. Most of the lesions regress with minor scarring; however, if nail dystrophy occurs, it is usually permanent.
The etiology of TBDN stems from weakly structured anchoring fibrils at the dermal-epidermal junction, causing a separation in the sublamina densa plane and leading to blister formation, which coincides with the pathophysiology of DEB. However, TBDN is distinguished by its self-limiting course and the presence of electron-dense inclusions, called stellate bodies. These appear within dilated rough endoplasmic reticulum of basal and suprabasal keratinocytes. Immunofluorescence and electron microscopy show that these inclusions contain type 7 collagen, the key structural protein in anchoring fibrils. Immunofluorescence reveals a granular pattern for type 7 collagen in the lower epidermis and a thinned linear pattern at the dermal-epidermal junction, which is considered a distinctive feature of TBDN. The clinical course of improvement seen in most TBDN cases reflects the eventual secretion of type 7 collagen from the keratinocytes to the epidermal basement membrane, correcting the structure of the anchoring fibrils and maintaining the dermal-epidermal junction.
The incidence of TBDN does not exhibit a difference in sex or race / ethnicity. While some cases of TBDN seem to be sporadic, studies have shown that it can be inherited in an autosomal dominant or recessive pattern as well as in a compound heterozygous fashion, in association with different mutations in the type 7 collagen gene COL7A1.
Transient bullous dermolysis of the newborn
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
Q81.2 – Epidermolysis bullosa dystrophica
SNOMEDCT:
254185007 – Epidermolysis bullosa dystrophica
Q81.2 – Epidermolysis bullosa dystrophica
SNOMEDCT:
254185007 – Epidermolysis bullosa dystrophica
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Updated:01/25/2022