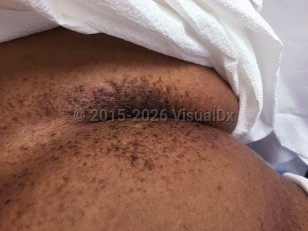
Galli-Galli disease (GGD) is a rare autosomal dominant genodermatosis that falls under the broader category of reticulate pigmented disorders of the skin. GGD is thought to be an allelic variant of Dowling-Degos disease (DDD), the latter a pigmentary disorder characterized by progressive hyperpigmentation of flexural skin. The distinguishing feature of GGD is the presence of suprabasal acantholysis histologically, and as such it has also been described as "acantholytic Dowling-Degos disease" in the literature.
In 1982, Bardach et al described the first cases of disease occurring in two brothers named Galli. The brothers presented with a reticulated hyperpigmented eruption affecting the skin folds. Biopsy demonstrated proliferation of rete ridges and basal layer hyperpigmentation as seen in DDD, but also focal suprabasal acantholysis.
Initially, GGD was favored to be a distinct clinical entity from DDD due to the unique and consistent feature of acantholysis; however, subsequent research supports a common etiology for DDD and GGD, as both have been found to share the same genetic defect.
Frameshift or nonsense KRT5 gene mutations lead to haploinsufficiency of keratin 5, a protein that plays an important role in cell-cell adhesion, epidermal differentiation, and melanosome uptake. The mechanism by which specific KRT5 mutations seen in DDD and GGD cause skin disease or correlate with variable histological subtypes is not yet elucidated.
Galli-Galli disease
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
L81.9 – Disorder of pigmentation, unspecified
SNOMEDCT:
370172004 – Skin pigmentation
L81.9 – Disorder of pigmentation, unspecified
SNOMEDCT:
370172004 – Skin pigmentation
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Updated:03/20/2018
Galli-Galli disease